doi.org/10.20986/revesppod.2025.1740/2025
REVISIÓN
Revisión sistemática de la afectación podológica en el síndrome de andrade
Systematic review of podiatric involvement in andrade syndrome
María del Pilar Alfageme García1
Belinda Basilio Fernández1
Manuel Martí Antonio2
Adela Gómez Luque3
Alba Torres Pére3
Sonia Hidalgo Ruiz1
1Centro Universitario de Plasencia. Universidad de Extremadura. España
2Departamento de Matemáticas. Universidad de Extremadura. España
3Centro Universitario de Cáceres. Universidad de Extremadura. España
Resumen
Introducción: El síndrome de Andrade, o polineuropatía amiloidótica familiar tipo I, es un trastorno genético raro causado por mutaciones en el gen de la transtiretina. Estas mutaciones provocan depósitos amiloides en los nervios periféricos y otros tejidos. Los pies se encuentran entre las regiones más afectadas, provocando pérdida sensorial, lesiones no reconocidas, infecciones y deformidades como el pie de Charcot. El presente trabajo realiza una revisión sistemática para determinar las complicaciones del pie en pacientes con síndrome de Andrade.
Material y métodos: Se realizó una revisión sistemática de acuerdo con las directrices PRISMA 2020. Se realizaron búsquedas en PubMed, Web of Science y Google Scholar hasta diciembre de 2024. Se incluyeron 13 estudios publicados entre 2019 y 2024. El riesgo de sesgo se evaluó mediante las herramientas ROBINS-I y RoB 2.
Resultados: Las complicaciones del pie en pacientes con síndrome de Andrade incluyeron úlceras, infecciones, deformidades estructurales, dolor neuropático y pie de Charcot. Estas afecciones se asociaron a una reducción de la movilidad, un aumento del riesgo de caídas y una disminución de la calidad de vida. Las estrategias terapéuticas incluían fisioterapia, ortesis, calzado especializado y tratamiento farmacológico precoz con tafamidis o patisirán.
Conclusión: Las complicaciones relacionadas con los pies son frecuentes en los pacientes con síndrome de Andrade y merman significativamente su independencia funcional y su bienestar. La detección precoz y la atención multidisciplinar son esenciales para mejorar los resultados clínicos y preservar la calidad de vida.
Palabras clave: Amiloidosis, polineuropatía, lesiones del pie, calidad de vida
Abstract
Introduction: Andrade syndrome, or familial amyloid polyneuropathy type I, is a rare genetic disorder caused by mutations in the transthyretin gene. These mutations lead to amyloid deposits in peripheral nerves and other tissues. The feet are among the most affected regions, resulting in sensory loss, unrecognized injuries, infections, and deformities such as Charcot foot. This paper makes a systematic review of the frequency of foot complications in patients with Andrade syndrome.
Materials and methods: A systematic review was conducted in accordance with PRISMA 2020 guidelines. Searches were performed in PubMed, Web of Science, and Google Scholar up to December 2024. Thirteen studies published between 2019 and 2024 were included. Risk of bias was assessed using ROBINS-I and RoB 2 tools.
Results: Foot complications in patients with Andrade syndrome included ulcers, infections, structural deformities, neuropathic pain, and Charcot foot. These conditions were associated with reduced mobility, increased risk of falls, and decreased quality of life. Therapeutic strategies included physiotherapy, orthoses, specialized footwear, and early pharmacological treatment with tafamidis or patisirán.
Conclusion: Foot-related complications are common in patients with Andrade syndrome and significantly impair their functional independence and well-being. Early detection and multidisciplinary care are essential to improve clinical outcomes and preserve quality of life.
Correspondencia
Belinda Basilio Fernández
bbasfer@unex.es
Recibido: 08-06-2025
Aceptado: 01-09-2025
Introducción
El síndrome de Andrade, también conocido como polineuropatía amiloidótica familiar tipo I (PAF tipo I), es un trastorno autosómico dominante poco frecuente causado por mutaciones en el gen de la transtiretina (TTR) (1,2). Estas mutaciones producen el mal plegamiento de la proteína transtiretina y la deposición de amiloide en diversos tejidos, con especial afinidad por los nervios periféricos, aunque la afectación cardiaca y gastrointestinal también es frecuente(3).
Clínicamente, la enfermedad se manifiesta como una polineuropatía sensitivo-motora distal, simétrica y progresiva, que suele iniciarse en los pies. La pérdida sensitiva, la debilidad muscular y la alteración del equilibrio hacen que el pie sea particularmente vulnerable desde fases tempranas. La neuropatía periférica resultante compromete la percepción del dolor, la presión y la temperatura, aumentando el riesgo de lesiones inadvertidas, infecciones persistentes y deformidades estructurales.
A diferencia de otras neuropatías periféricas, como la neuropatía diabética o la enfermedad de Charcot-Marie-Tooth, el síndrome de Andrade suele presentar una disfunción autonómica y sistémica más extendida. Esta combinación incrementa la probabilidad de complicaciones podológicas graves, como inestabilidad articular, úlceras crónicas y artropatías destructivas similares al pie de Charcot. Aunque se describen con menor frecuencia, estas complicaciones pueden afectar de manera significativa la marcha, el equilibrio y la independencia del paciente(4,5). Además, el compromiso autonómico reduce la regulación vascular y la integridad cutánea, retrasando la cicatrización de heridas y aumentando la susceptibilidad a las úlceras plantares5. El dolor neuropático puede aparecer de forma temprana, incluso antes de los síntomas motores, deteriorando progresivamente el bienestar físico y emocional(6,7).
Pese a los avances terapéuticos —como tafamidis y patisirán, que han mostrado eficacia para frenar el avance de la enfermedad(2,8)—, las consecuencias podológicas del síndrome están poco documentadas. La mayor parte de la literatura disponible se centra en aspectos genéticos y neurológicos, con escasa atención a las implicaciones?funcionales de las lesiones en el pie o a la evaluación de estrategias terapéuticas y preventivas(9). Por ello, esta revisión sistemática tiene como objetivo evaluar la prevalencia y las características clínicas de las complicaciones podológicas en pacientes con síndrome de Andrade, analizar cómo estas afectan la movilidad y la calidad de vida, e identificar las lesiones más frecuentes, los factores de riesgo asociados y las intervenciones terapéuticas actualmente aplicadas, junto con su eficacia clínica y viabilidad.
Materiales y métodos
Esta revisión sistemática se realizó de conformidad con las guías PRISMA 2020 (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) (10,11)y fue registrada prospectivamente en la base de datos PROSPERO con el identificador CRD581310. Entre los estudios incluidos se encuentra el ensayo NEURO-Transform de Coelho y cols. (12), un estudio fase 3 abierto que proporciona datos demográficos y clínicos detallados de pacientes con polineuropatía asociada a amiloidosis hereditaria por transtiterina (ATTRv). Se seleccionó por su exhaustiva caracterización de la progresión neuropática y de los resultados terapéuticos, aspectos relevantes para las implicaciones podológicas analizadas.
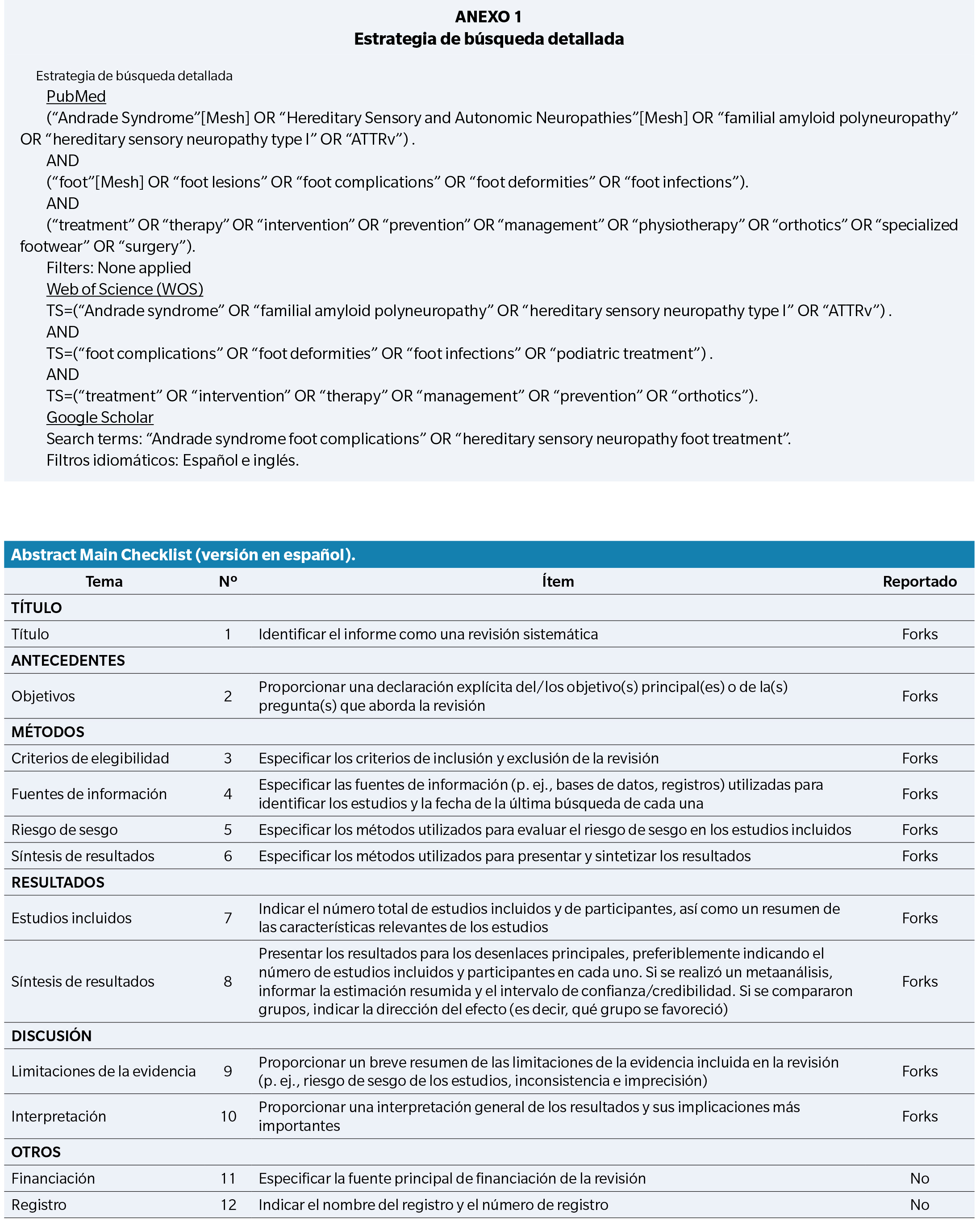
Estrategia de búsqueda
Se realizó una búsqueda sistemática en las bases de datos PubMed, Google Scholar y Web of Science (WOS) hasta el 31 de diciembre de 2024. Google Scholar se incluyó para identificar literatura gris y fuentes no indexadas en bases tradicionales.
Se utilizaron las siguientes palabras clave: “Andrade syndrome”, “Hereditary sensory neuropathy type I”, “foot lesions”, “foot complications”, “foot deformities”, “foot infections”, “treatment”, “therapy”, “management”, “intervention”, “prevention”, “specialized footwear”, “physiotherapy”, “surgery”, “orthotics”, “medications”.
Se aplicaron tanto términos MeSH como operadores booleanos (AND, OR) para combinar la búsqueda. Las cadenas se adaptaron a cada base de datos.
Se incluyeron artículos con resúmenes y textos completos en inglés o español, sin límite de año ni filtros adicionales.
La estrategia completa de búsqueda para cada base se detalla en el Anexo 1.
Criterios de inclusión y exclusión
Los criterios de inclusión fueron estudios primarios publicados en revistas revisadas por pares indexadas en PubMed, ScienceDirect o Google Scholar; estudios que investiguen el impacto del síndrome de Andrade/ATTRv en la salud del pie; estudios que evalúen complicaciones, movilidad, calidad de vida o intervenciones preventivas/terapéuticas; y publicaciones entre 2019 y 2024. Estudios en inglés o español.
Los criterios de exclusión fueron revisiones, editoriales, cartas al editor, resúmenes de congresos u opiniones. Estudios que no aborden específicamente el síndrome de Andrade o sus implicaciones podológicas. Publicaciones previas a 2019 o en otros idiomas distintos del inglés y el español. Trabajos que no cumplieran estándares metodológicos de calidad.
Proceso de selección
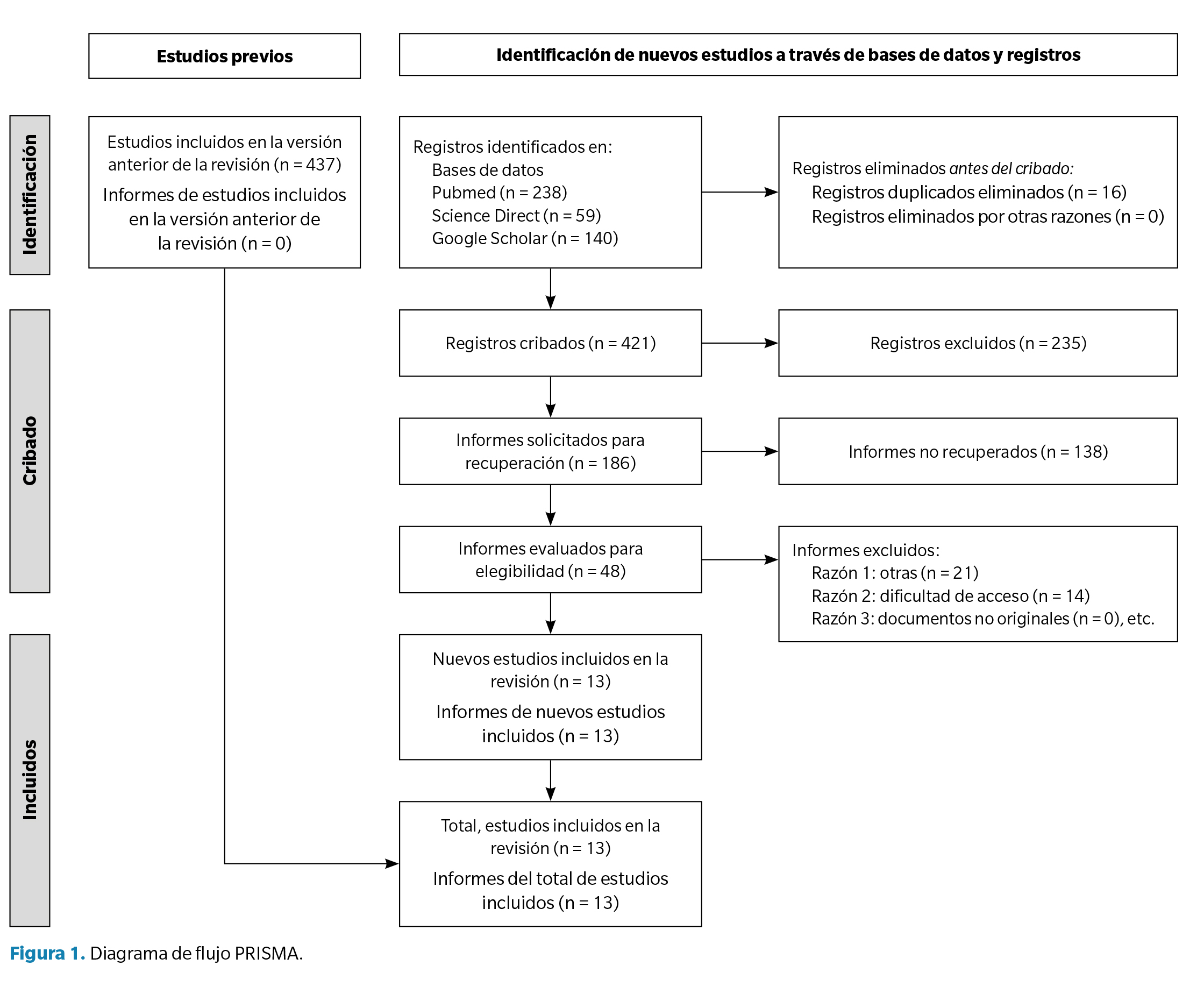
Todos los registros fueron exportados a un gestor bibliográfico para eliminar duplicados. Dos revisores independientes examinaron títulos y resúmenes; los textos completos fueron evaluados según los criterios establecidos. Los desacuerdos se resolvieron por consenso o con un tercer revisor. Un diagrama PRISMA (Figura 1) resume el proceso de selección.
Extracción y síntesis de datos
Los datos de los estudios seleccionados se volcaron en una tabla estandarizada (autores, año, diseño, características de la población, intervención/exposición, resultados principales).
Se aplicó una síntesis narrativa debido a la heterogeneidad de diseños y variables. Los resultados se agruparon por tipo de complicación podológica (lesiones, infecciones, deformidades), limitaciones funcionales e impacto en calidad de vida.
Evaluación del riesgo de sesgo
La calidad metodológica se evaluó con:
- ROBINS-I para estudios no aleatorizados.
- RoB 2 para ensayos clínicos aleatorizados.
Dos revisores llevaron a cabo la evaluación de forma independiente, resolviendo discrepancias mediante discusión.
Resultados
El diagrama que ilustra el proceso de selección de estudios en las distintas etapas de la revisión sistemática se muestra en la Figura 1. De un total de 499 estudios obtenidos en la búsqueda inicial, 13 documentos cumplieron los criterios de inclusión y fueron seleccionados: 3 estudios observacionales, 3 ensayos clínicos, un estudio retrospectivo, un consenso de expertos, una cohorte retrospectiva y un estudio descriptivo transversal. Además, se incorporaron 2 documentos adicionales a partir de la búsqueda manual en listas de referencias. Se compiló una tabla de artículos originales con las principales características de cada estudio (Tabla 1). Aunque se incluyeron trece documentos en la síntesis final, solo doce correspondieron a estudios originales con datos empíricos, mientras que el consenso de expertos se consideró relevante por sus aportaciones clínicas.
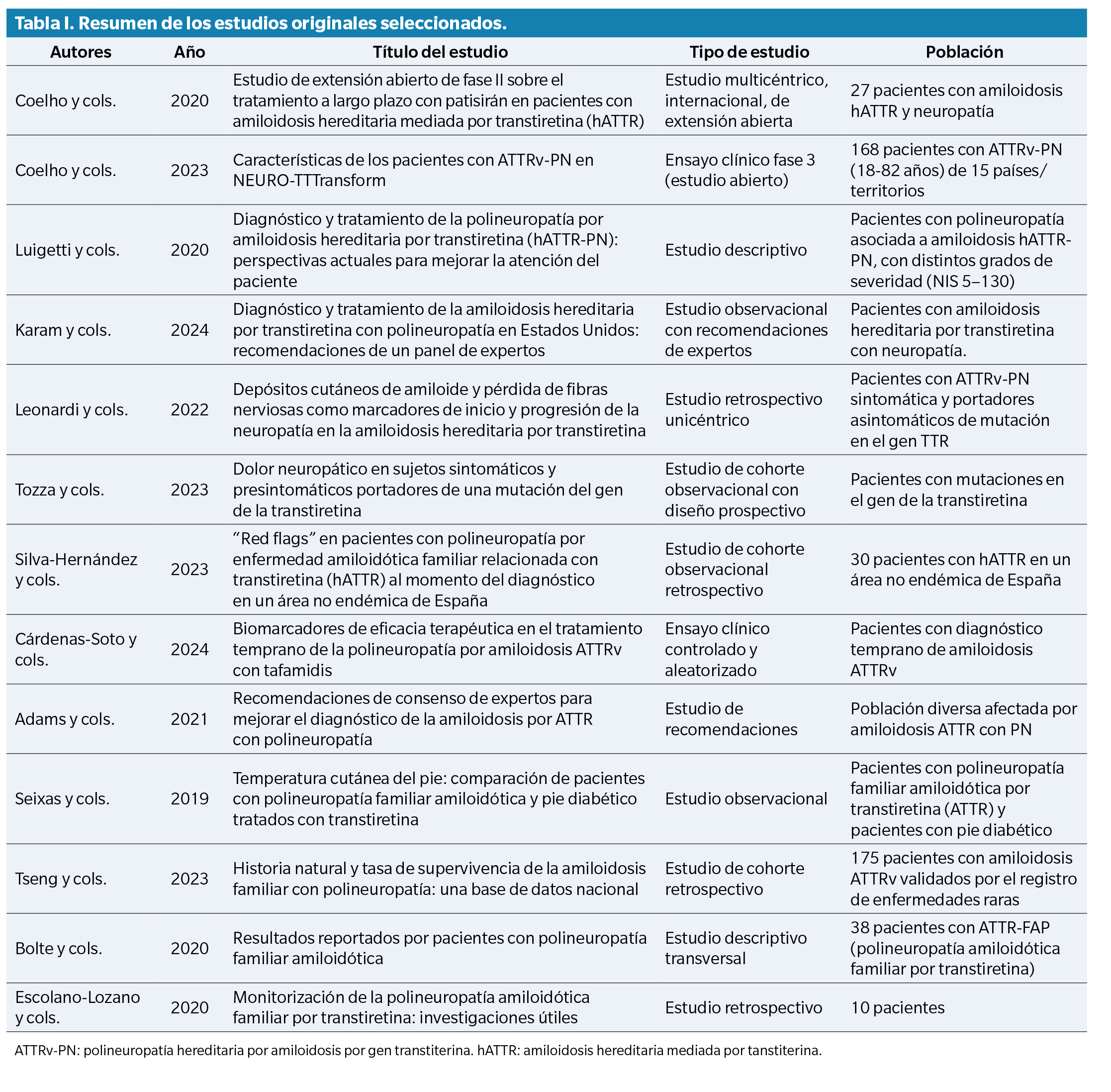
Entre los estudios seleccionados, varios informaron específicamente sobre signos podológicos de la ATTRv, incluidas úlceras plantares, deformidades biomecánicas y alteraciones de la termorregulación en los pies. Estas complicaciones son especialmente relevantes en la práctica podológica, ya que aumentan el riesgo de lesiones por presión, disfunción de la marcha y pérdida de autonomía.
Aunque el artículo de Karam y cols. se incluyó en la síntesis narrativa debido a su relevancia clínica, no fue evaluado mediante las herramientas ROBINS-I ni RoB 2, ya que no presenta datos originales de intervención. En su lugar, corresponde a un consenso de expertos basado en paneles cuyo objetivo es ofrecer recomendaciones clínicas, por lo que queda fuera del alcance de las herramientas de evaluación de sesgo aplicadas en esta revisión(13).
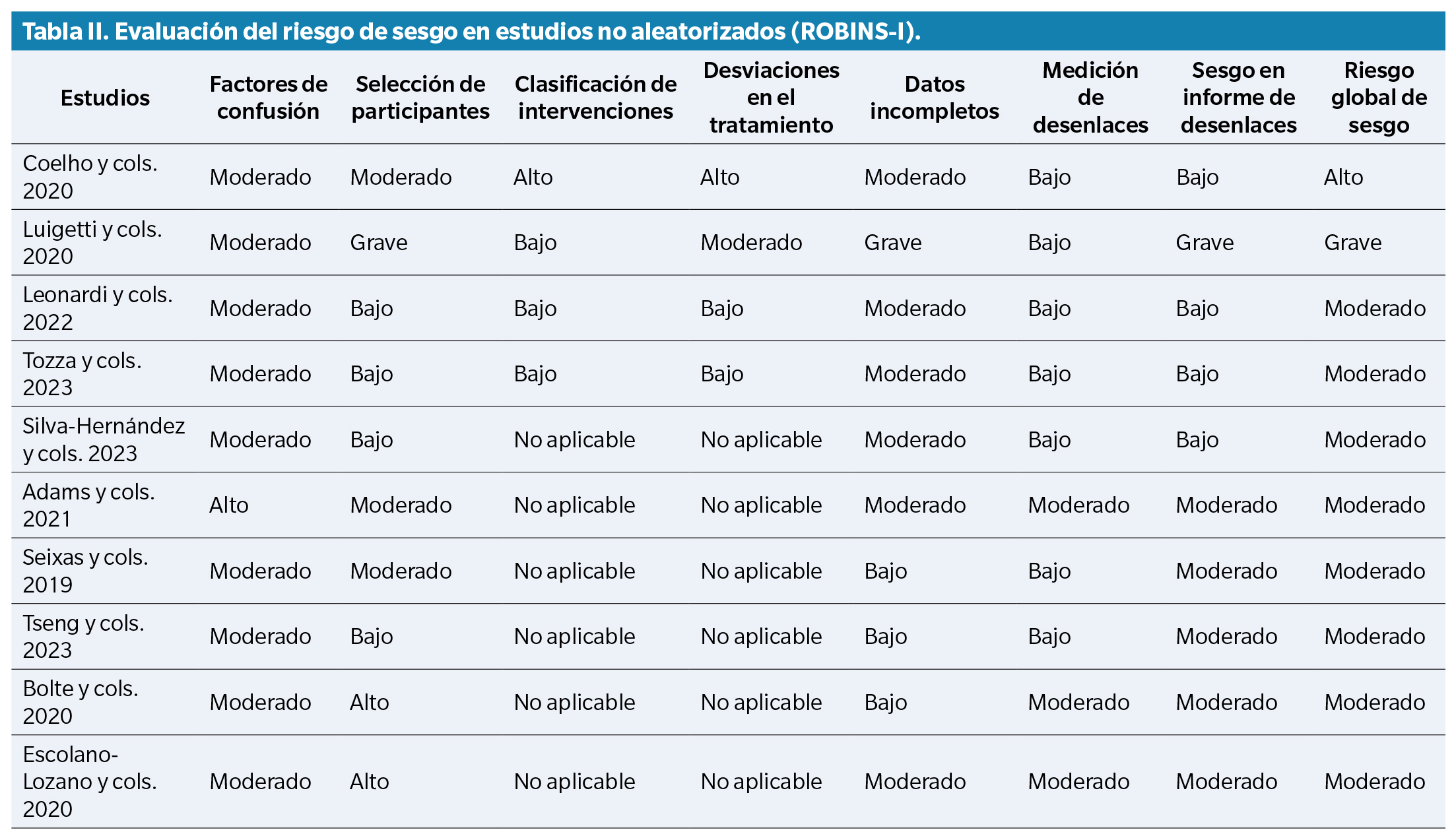
La evaluación del riesgo de sesgo se realizó utilizando 2 herramientas: ROBINS-I para los estudios no aleatorizados (Tabla 2) y RoB 2 para los estudios aleatorizados (Tabla 3).
Entre los estudios seleccionados, varios informaron específicamente sobre manifestaciones podológicas de la ATTRv, incluidas úlceras plantares, deformidades biomecánicas y alteraciones de la termorregulación en los pies. Además, Leonardi y cols. demostraron que los depósitos de amiloide en la piel se asocian con pérdida de fibras nerviosas en pacientes con ATTRv, lo cual sugiere su potencial valor como marcadores tempranos de progresión de la neuropatía(14). Silva-Hernández y cols. subrayaron la presencia de signos clínicos de alarma en el diagnóstico de pacientes de áreas no endémicas, lo que pone de relieve el riesgo de identificación tardía(15). Por su parte, Cárdenas-Soto y cols. evaluaron biomarcadores cutáneos que podrían guiar decisiones terapéuticas tempranas y mejorar la monitorización de la progresión de la enfermedad, con implicaciones directas para la salud del pie16. Estas complicaciones son especialmente relevantes para la práctica podológica, ya que aumentan el riesgo de lesiones por presión, disfunción de la marcha y pérdida de autonomía.

La polineuropatía amiloidótica familiar (ATTR) se correlaciona con una prevalencia significativa de complicaciones relacionadas con el pie, incluidas la neuropatía y las alteraciones de la temperatura cutánea, que pueden provocar vasoconstricción y disminución de la percepción sensorial(17,18,). Aunque se observan complicaciones similares en neuropatías diabéticas y de Charcot-Marie-Tooth, la naturaleza sistémica y la afectación autonómica de la ATTRv hacen que su impacto podológico sea más insidioso y con frecuencia infradiagnosticado. Esto refuerza la necesidad de estrategias específicas de cuidado del pie dirigidas a esta población.
En personas con diagnóstico de ATTR, la disminución observada de la inervación simpática se ha relacionado con el agravamiento de estas complicaciones, en particular en pacientes con diabetes mellitus concomitante(19). Un estudio de Leonardi y cols. (14) reveló que el 80 % de los individuos sintomáticos con neuropatía asociada a amiloidosis hereditaria presentaban depósitos amiloides en la dermis, lo cual podría contribuir a la degeneración de fibras nerviosas y, en consecuencia, a complicaciones como lesiones y deformidades en el pie. Además, la investigación de Karam y cols. (13) destaca que la neuropatía autonómica, que puede manifestarse como alteraciones de la función gastrointestinal y síntomas asociados, también afecta de manera negativa la salud del pie, generando complicaciones adicionales. Asimismo, Tseng y cols. (20) documentaron que los individuos con ATTRv tenían una mayor incidencia de complicaciones podológicas en comparación con quienes no presentaban la enfermedad, lo que subraya la necesidad de un seguimiento estrecho.
Las investigaciones han demostrado que la movilidad se encuentra significativamente comprometida en las personas diagnosticadas con ATTR, lo que repercute de manera negativa en su calidad de vida global3. Un estudio realizado por Coelho y cols. (21) evaluó específicamente el impacto de la neuropatía en la calidad de vida de los pacientes con PAF. Los resultados revelaron que los pacientes experimentan un deterioro considerable de la calidad de vida, asociado con manifestaciones como dolor neuropático, debilidad muscular y problemas de movilidad. Asimismo, se observó que la neuropatía autonómica, que puede manifestarse a través de síntomas gastrointestinales y cardiovasculares, también desempeña un papel en este deterioro. En un análisis transversal descriptivo, Bolte y cols. (18) evidenciaron que los pacientes con polineuropatía amiloidótica familiar reportaban una calidad de vida reducida relacionada con la disminución de la movilidad y el malestar podológico. Además, Karam y cols. (13) señalan que la confirmación diagnóstica de ATTR puede lograrse mediante biopsias e imágenes, lo que facilita un tratamiento más eficaz y, potencialmente, una mejor calidad de vida. El ensayo clínico aleatorizado de Lepesis y cols(19). mostró que las movilizaciones de pie y tobillo, junto con estiramientos domiciliarios, mejoraron la movilidad en pacientes con neuropatía diabética, lo que sugiere que estrategias análogas podrían ser beneficiosas en individuos con ATTR. La evaluación de la función autonómica mediante instrumentos como Sudoscan ha demostrado ser útil para delimitar alteraciones sensoriales en los pies, intrínsecamente relacionadas con la calidad de vida de estos pacientes(2,18).
Coelho y cols. (21) concluyeron que intervenciones terapéuticas adecuadas podrían tener un efecto favorable sobre la progresión de la enfermedad y mejorar la calidad de vida de los pacientes con PAF. Se enfatizó la importancia del diagnóstico temprano y el tratamiento oportuno para optimizar los resultados terapéuticos. Tozza y cols.6 resaltaron la elevada prevalencia y la aparición temprana del dolor neuropático en individuos con mutaciones del gen TTR. Este estudio de 2023 mostró que el dolor está presente con frecuencia incluso en portadores presintomáticos, lo que respalda la necesidad de evaluaciones sensoriales precoces y cuidados podológicos preventivos. El dolor neuropático puede restringir la capacidad de los pacientes para realizar actividades físicas y, en consecuencia, reducir su movilidad global. La restricción del movimiento libre no solo limita la autonomía física, sino que también puede contribuir a un ciclo de inactividad que agrava los síntomas neuropáticos y la debilidad muscular. La calidad de vida de los pacientes se ve comprometida no solo por el malestar físico, sino también por las limitaciones de movilidad. La imposibilidad de realizar actividades cotidianas, laborales o recreativas puede generar frustración, aislamiento y depresión. Diversos estudios postulan que un tratamiento eficaz del dolor neuropático es esencial para mejorar la calidad de vida, ya que el alivio del dolor permite a los pacientes recuperar cierto grado de movilidad y, con ello, su independencia y bienestar general.
La investigación de Silva-Hernández y cols. (15) aborda las implicaciones de la neuropatía asociada con la amiloidosis hereditaria en la movilidad de los pacientes. Signos como la debilidad muscular y las alteraciones del equilibrio pueden obstaculizar la capacidad de desplazarse de manera segura y eficiente. Esto no solo incrementa el riesgo de caídas y lesiones posteriores, sino que también conlleva una mayor dependencia de otras personas para la realización de tareas rutinarias, lo cual compromete la autonomía del paciente. La calidad de vida se ve profundamente influida por las limitaciones de movilidad. La investigación pone de manifiesto que la calidad de vida de estos individuos no solo se evalúa desde la perspectiva de la salud física, sino también a través de su capacidad de participar en interacciones sociales y actividades familiares. La movilidad constituye un elemento fundamental de la calidad de vida, y su deterioro puede tener efectos adversos en el bienestar emocional y social de los pacientes.
Las modalidades terapéuticas incluyen movilizaciones de pie y tobillo junto con ejercicios de estiramiento, que han demostrado resultados favorables en cuanto a la movilidad en pacientes con neuropatía diabética periférica(19). Asimismo, se postula que la expansión de investigaciones centradas en intervenciones terapéuticas específicas podría mejorar la atención de los pacientes y mitigar las complicaciones podológicas(2.3).
El estudio de Seixas y cols. (5), titulado “Efectos de la amiloidosis hereditaria por transtiretina en la calidad de vida de los pacientes”, examina el impacto de la amiloidosis hereditaria por transtiretina (AHTT) en la calidad de vida de los afectados. Los principales hallazgos indican que los individuos con AHTT experimentan un deterioro sustancial de su calidad de vida, atribuible a síntomas asociados con la enfermedad, incluidos dolor neuropático, debilidad muscular y disfunción autonómica. El análisis identifica síntomas prevalentes que afectan negativamente la calidad de vida, como los trastornos gastrointestinales, las alteraciones de la movilidad y los signos cardiovasculares. Estos síntomas no solo repercuten en la salud física, sino que también tienen consecuencias emocionales y sociales. En base a los informes de los pacientes, se establece una asociación significativa entre la gravedad de los síntomas y la calidad de vida. A medida que aumenta la intensidad de los síntomas, se observa un deterioro concomitante de la calidad de vida, lo que pone de relieve la necesidad de un manejo sintomático eficaz. La investigación enfatiza la importancia de un tratamiento oportuno y adecuado para mejorar la calidad de vida de los pacientes. Se propone un enfoque holístico de la atención, que combine intervenciones farmacológicas con apoyo psicológico y social, como elemento esencial para mejorar el bienestar global de los pacientes. El estudio(6) aclara los efectos profundos de la amiloidosis hereditaria por transtiretina en la vida cotidiana de los pacientes, lo cual pone de manifiesto la necesidad de estrategias multidisciplinarias en el tratamiento de la enfermedad, que deben centrarse no solo en los aspectos físicos, sino también en las dimensiones emocionales y sociales relevantes para la calidad de vida.
La investigación de Cárdenas-Soto y cols. (16) se centró en evaluar la eficacia terapéutica de tafamidis en individuos con diagnóstico preliminar de amiloidosis hereditaria por transtiretina (ATTRv). Este estudio indica que la administración de tafamidis en fases tempranas de la polineuropatía amiloidótica puede mejorar los resultados clínicos y resalta la necesidad de seguir explorando biomarcadores que faciliten la personalización del tratamiento y la monitorización de la eficacia terapéutica. Esta investigación revela que la intervención con tafamidis es eficaz para ralentizar la progresión de la neuropatía en comparación con una cohorte control. Este hallazgo implica que tafamidis podría constituir una opción terapéutica beneficiosa para pacientes en fases iniciales de la enfermedad. Los biomarcadores cutáneos también se estudian como posibles indicadores de eficacia terapéutica. Estos biomarcadores pueden aportar información sobre la respuesta al tratamiento y la progresión de la enfermedad, elementos vitales para la monitorización clínica de la amiloidosis.El estudio subraya el papel fundamental del diagnóstico temprano de la ATTRv, ya que una intervención oportuna con tafamidis puede influir de manera significativa tanto en la calidad de vida como en la trayectoria de la progresión de la enfermedad.
La investigación de Escolano-Lozano y cols. (17), titulada “Evaluación de la neuropatía en pacientes con amiloidosis hereditaria por transtiretina: un enfoque multidimensional”, se centra en la valoración y el tratamiento de la neuropatía en pacientes con diagnóstico de ATTRv.Los autores defienden la necesidad de un abordaje multidimensional en la evaluación de la neuropatía, que incorpore tanto valoraciones clínicas como el uso de herramientas diagnósticas sofisticadas. Esta metodología facilita una caracterización más completa de la neuropatía en estos pacientes.
El estudio de Escolano-Lozano y cols. (17) aclara la compleja naturaleza de la neuropatía en la amiloidosis hereditaria por transtiretina y la necesidad de un abordaje holístico en su evaluación y tratamiento. Al abordar las dimensiones clínicas y funcionales de la neuropatía, se puede mejorar la calidad de vida de los pacientes y optimizar los resultados terapéuticos.
Estos estudios avalan el objetivo inicial de esta revisión sistemática: determinar en qué medida las complicaciones podológicas en pacientes con ATTRv afectan la movilidad y la calidad de vida. La evidencia pone de relieve la necesidad de una participación multidisciplinaria podológica, particularmente en la identificación temprana y el manejo del dolor neuropático, las deformidades y la disfunción autonómica en las extremidades inferiores.
Discusión
Esta revisión sistemática demuestra que el síndrome de Andrade (PAF tipo I) tiene un impacto significativo en la salud del pie, comparable en gravedad —aunque no en prevalencia— al de otras neuropatías más estudiadas, como la diabetes mellitus o la enfermedad de Charcot-Marie-Tooth(4,7,10). La pérdida sensitiva progresiva, junto con la disfunción autonómica característica, incrementa la vulnerabilidad a lesiones, ulceraciones e infecciones, comprometiendo directamente la calidad de vida y la independencia funcional(5,14,17).
A diferencia de la neuropatía diabética, donde la incidencia de pie de Charcot o de úlceras plantares está ampliamente documentada, las complicaciones podológicas en el síndrome de Andrade permanecen insuficientemente descritas6,10. Sin embargo, estudios recientes confirman que los déficits sensitivos y la infiltración amiloide en la dermis y fibras nerviosas pueden originar alteraciones biomecánicas de magnitud similar(12,14,15).
La mayoría de los estudios coinciden en señalar un deterioro progresivo de la movilidad, especialmente en fases avanzadas, lo cual afecta de manera significativa la capacidad de realizar actividades de la vida diaria(13,18,20). Dicho deterioro ha sido medido mediante escalas de calidad de vida (EQ-5D, Norfolk QoL-DN), revelando altos niveles de discapacidad frente a otras neuropatías hereditarias(11,18,21). Además, la carga psicológica, incluido el miedo a las lesiones y el aislamiento social, añade una complejidad adicional al cuidado de los pacientes.
Aunque terapias modificadoras como tafamidis y patisirán han demostrado eficacia para ralentizar la progresión neurológica(2,8,16), la literatura revisada evidencia una falta de estrategias centradas en la prevención y el tratamiento de complicaciones podológicas. Intervenciones como fisioterapia, ortesis personalizadas, educación en salud podológica y manejo específico del dolor podrían desempeñar un papel esencial en la atención integral(5,13,19).
El pie se perfila como una de las estructuras anatómicas más afectadas en el síndrome de Andrade debido a la distribución distal de la neuropatía. Esta revisión subraya la necesidad de integrar la evaluación podológica especializada como un componente esencial del manejo integral de la enfermedad. La atención multidisciplinaria debe incluir no solo estrategias dirigidas a frenar la progresión neurológica, sino también medidas para prevenir complicaciones estructurales, preservar la movilidad y mantener la autonomía funcional. Reforzar este enfoque clínico podría mejorar los resultados terapéuticos y la calidad de vida global de las personas afectadas por esta condición(2,8,12,13,19).
La principal limitación de esta revisión es el número reducido de estudios que abordan de forma directa la afectación del pie en el síndrome de Andrade. Muchos artículos se centran en aspectos neurológicos o genéticos generales, sin incluir variables podológicas específicas. La heterogeneidad metodológica, la variabilidad en las medidas de resultado y el tamaño reducido de las muestras limitan la posibilidad de síntesis cuantitativa firme.
Las recomendaciones extraíbles de la presente revisión sistemática hacen referencia a desarrollar estudios clínicos centrados en el pie como unidad funcional clave en el avance de la discapacidad en pacientes con ATTRv, incorporar de forma sistemática la evaluación podológica en el seguimiento multidisciplinario de la enfermedad, investigar la eficacia de intervenciones como movilización articular, calzado ortopédico o sensores termográficos para la detección precoz de lesiones5,13,19 y establecer protocolos de cribado podológico, especialmente en áreas no endémicas, donde el diagnóstico suele demorarse(15,16).
Conclusiones
Esta revisión sistemática confirma que la polineuropatía amiloidótica familiar (PAF), también conocida como amiloidosis hereditaria por transtiretina (ATTRv), tiene un impacto notable en la salud del pie, debido principalmente a la neuropatía sensitiva y autonómica progresiva. Los pacientes experimentan una marcada reducción en la percepción de dolor y temperatura en las extremidades distales, lo que aumenta el riesgo de lesiones inadvertidas, ulceraciones y deformidades biomecánicas.
Estas complicaciones afectan de manera severa la marcha, el equilibrio y la autonomía funcional, contribuyendo a la pérdida progresiva de movilidad y al deterioro significativo de la calidad de vida. El dolor neuropático y la cicatrización retardada agravan la discapacidad, sobre todo en fases avanzadas de la enfermedad.
Si bien los tratamientos modificadores, como tafamidis y patisirán, han demostrado ser eficaces a la hora de frenar la progresión neurológica, la literatura evidencia una clara carencia de estrategias orientadas a la prevención y tratamiento de las complicaciones podológicas. Pocos estudios han abordado intervenciones como ortesis, fisioterapia o educación en autocuidado, a pesar de su potencial beneficio clínico.
Es fundamental desarrollar un modelo asistencial que integre la evaluación podológica especializada como parte del seguimiento multidisciplinario. Asimismo, se requieren más investigaciones que evalúen la eficacia de intervenciones específicas dirigidas a preservar la funcionalidad, reducir la morbilidad podológica y mejorar la calidad de vida de los pacientes con síndrome de Andrade.
Conflictos de intereses
Los autores declaran que no existen conflictos de intereses relacionados con la realización y publicación de este estudio.
Fuentes de financiación
Ninguna.
Contribución de los autores
Concepción y diseño del estudio: MPAG, ATG, SHR. Búsqueda bibliográfica/recopilación y selección de estudios: MPAG, BBF, SHR. Análisis e interpretación de resultados: BBF, MMA. Elaboración, redacción y preparación del borrador: AGL, BBF, MPAG.Revisión final: SHR, BBF, MPAG.
Material suplementario
Anexo 1
Bibliografía
- Murakami T, Yokoyama T, Mizuguchi M, Toné S, Takaku S, Sango K, et al. A low amyloidogenic E61K transthyretin mutation may cause familial amyloid polyneuropathy. J Neurochem. 2021;156(6):957-66. DOI: 10.1111/jnc.15162.
- Luigetti M, Romano A, di Paolantonio A, Bisogni G, Sabatelli M. Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: Current perspectives on improving patient care. Ther Clin Risk Manag. 2020;16:109-23. DOI:10.2147/TCRM.S219979.
- Adams D, Ando Y, Beirão JM, Coelho T, Gertz MA, Gillmore JD, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109-22. DOI:10.1007/s00415-019-09688-0.
- Kucera T, Shaikh HH, Sponer P. Charcot neuropathic arthropathy of the foot: A literature review and single-center experience. J Diabetes Res. 2016;2016:3207043. DOI:10.1155/2016/3207043.
- Seixas A, Vilas-Boas MC, Carvalho R, Coelho T, Ammer K Vilas-Boas JP, et al. Skin temperature of the foot: Comparing transthyretin familial amyloid polyneuropathy and diabetic foot patients. Comput Methods Biomech Biomed Eng Imaging Vis. 2019;7(5-6):504-11. DOI:10.1080/21681163.2018.1471621.
- Tozza S, Luigetti M, Antonini G, Mazzeo A Severi D, di Paolantonio A, et al. Neuropathic pain experience in symptomatic and presymptomatic subject carrying a transthyretin gene mutations. Front Neurol. 2023;14:1109782. DOI:10.3389/fneur.2023.1109782.
- Girach A, Julian TH, Varrassi G, Paladini A, Vadalouka A, Zis P. Quality of life in painful peripheral neuropathies: A systematic review. Pain Res Manag. 2019;2019:2091960. DOI:10.1155/2019/2091960.
- Coelho T, Maia LF, Martins da Silva A, Cruz MW, Palnté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatmen of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14. DOI: 10.1007/s00415-013-7051-7.
- Ziegler D, Burow S, Landgraf R, Lobmann R, Reiners K, Rett K, et al. Current practice of podiatrists in testing for diabetic polyneuropathy and implementing foot care (PROTECT Study Survey 2). Endocr Pract. 2024;30(9):817-21. DOI:10.1016/j.eprac.2024.06.006.
- Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. DOI:10.1371/journal.pmed.1000097.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. DOI:10.1136/bmj.n71.
- Coelho T, Waddington Cruz M, Chao CC, Parman Y, Wixner J, Weiler M, et al. Characteristics of patients with hereditary transthyretin amyloidosis-polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an open-label phase 3 study of eplontersen. Neurol Ther. 2023;12(1):267-87. DOI: 10.1007/s40120-022-00414-z.
- Karam C, Mauermann ML, Gonzalez-Duarte A, Kaku MC, Ajroud-Driss S, Brannagan TH, et al. Diagnosis and treatment of hereditary transthyretin amyloidosis with polyneuropathy in the United States: Recommendations from a panel of experts. Muscle Nerve. 2024;69(3):273-87. DOI: 10.1002/mus.28026.
- Leonardi L, Adam C, Beaudonnet G, Beauvais D, Cauquil C, Not A, et al. Skin amyloid deposits and nerve fiber loss as markers of neuropathy onset and progression in hereditary transthyretin amyloidosis. Eur J Neurol. 2022;29(5):1477-87. DOI: 10.1111/ene.15268.
- Silva-Hernández L, Horga Hernández A, Valls Carbó A, Guerrero Sola A, Montalvo-Moraleda MT, Galán Dávila L. Red flags in patients with hereditary transthyretin amyloidosis at diagnosis in a non-endemic area of Spain. Neurology (Eng Ed). 2023;38(2):87-92. DOI: 10.1016/j.nrl.2020.06.009.
- Cárdenas-Soto K, Dominguez X, Cortes G, Tsai F, Saniger MDM, Guraieb-Chahin P, et al. Cutaneous biomarkers of therapeutic efficacy in early treatment of hereditary ATTR amyloid polyneuropathy with tafamidis. J Peripheral Nervous Sys. 2024;29(2):221-31. DOI: 10.1111/jns.12624.
- Escolano-Lozano F, Geber C, Barreiros AP, Birklein F. Follow-up in transthyretin familial amyloid polyneuropathy: Useful investigations. J Neurol Sci. 2020; 413:116776. DOI: 10.1016/j.jns.2020.116776.
- Bolte FJ, Langenstroer C, Friebel F, Hüsing-Kabar A, Dugas M, Schmidt HH. Patient-reported outcomes on familial amyloid polyneuropathy (FAP). Orphanet J Rare Dis. 2020;15(1):287. DOI: 10.1186/s13023-020-01575-6.
- Lepesis V, Paton J, Rickard A, Latour JM, Marsden J. Effects of foot and ankle mobilisations combined with home stretches in people with diabetic peripheral neuropathy: A proof-of-concept RCT. J Foot Ankle Res. 2023;16(1):88. DOI: 10.1186/s13047-023-00690-4.
- Tseng W, Huang H, Li C, Chang C, Chan WP, Lin K, et al. Natural history and survival rate of familial amyloidosis with polyneuropathy: A nationwide databank. Ann Clin Transl Neurol. 2023;10(5):779-86. DOI: 10.1002/acn3.51765.
- Coelho T, Adams D, Conceição I, Waddington-Cruz M, Schmidt HH, Buades J, et al. A phase II, open-label, extension study of long-term patisirán treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. DOI: 10.1186/s13023-020-01399-4.
Anexo