doi.org/10.20986/revesppod.2025.1740/2025
REVIEW
Systematic review of podiatric involvement in andrade syndrome
Revisión sistemática de la afectación podológica en el síndrome de andrade
María del Pilar Alfageme García1
Belinda Basilio Fernández1
Manuel Martí Antonio2
Adela Gómez Luque3
Alba Torres Pére3
Sonia Hidalgo Ruiz1
1Centro Universitario de Plasencia. Universidad de Extremadura. España
2Departamento de Matemáticas. Universidad de Extremadura. España
3Centro Universitario de Cáceres. Universidad de Extremadura. España
Abstract
Introduction: Andrade syndrome, or familial amyloid polyneuropathy type I, is a rare genetic disorder caused by mutations in the transthyretin gene. These mutations lead to amyloid deposits in peripheral nerves and other tissues. The feet are among the most affected regions, resulting in sensory loss, unrecognized injuries, infections, and deformities such as Charcot foot. This paper makes a systematic review of the frequency of foot complications in patients with Andrade syndrome.
Materials and methods: A systematic review was conducted in accordance with PRISMA 2020 guidelines. Searches were performed in PubMed, Web of Science, and Google Scholar up to December 2024. Thirteen studies published between 2019 and 2024 were included. Risk of bias was assessed using ROBINS-I and RoB 2 tools.
Results: Foot complications in patients with Andrade syndrome included ulcers, infections, structural deformities, neuropathic pain, and Charcot foot. These conditions were associated with reduced mobility, increased risk of falls, and decreased quality of life. Therapeutic strategies included physiotherapy, orthoses, specialized footwear, and early pharmacological treatment with tafamidis or patisirán.
Conclusion: Foot-related complications are common in patients with Andrade syndrome and significantly impair their functional independence and well-being. Early detection and multidisciplinary care are essential to improve clinical outcomes and preserve quality of life.
Correspondencia
Belinda Basilio Fernández
bbasfer@unex.es
Resumen
Introducción: El síndrome de Andrade, o polineuropatía amiloidótica familiar tipo I, es un trastorno genético raro causado por mutaciones en el gen de la transtiretina. Estas mutaciones provocan depósitos amiloides en los nervios periféricos y otros tejidos. Los pies se encuentran entre las regiones más afectadas, provocando pérdida sensorial, lesiones no reconocidas, infecciones y deformidades como el pie de Charcot. El presente trabajo realiza una revisión sistemática para determinar las complicaciones del pie en pacientes con síndrome de Andrade.
Material y métodos: Se realizó una revisión sistemática de acuerdo con las directrices PRISMA 2020. Se realizaron búsquedas en PubMed, Web of Science y Google Scholar hasta diciembre de 2024. Se incluyeron 13 estudios publicados entre 2019 y 2024. El riesgo de sesgo se evaluó mediante las herramientas ROBINS-I y RoB 2.
Resultados: Las complicaciones del pie en pacientes con síndrome de Andrade incluyeron úlceras, infecciones, deformidades estructurales, dolor neuropático y pie de Charcot. Estas afecciones se asociaron a una reducción de la movilidad, un aumento del riesgo de caídas y una disminución de la calidad de vida. Las estrategias terapéuticas incluían fisioterapia, ortesis, calzado especializado y tratamiento farmacológico precoz con tafamidis o patisirán.
Conclusión: Las complicaciones relacionadas con los pies son frecuentes en los pacientes con síndrome de Andrade y merman significativamente su independencia funcional y su bienestar. La detección precoz y la atención multidisciplinar son esenciales para mejorar los resultados clínicos y preservar la calidad de vida.
Palabras clave: Amiloidosis, polineuropatía, lesiones del pie, calidad de vida
Corresponding author
Belinda Basilio Fernández
bbasfer@unex.es
Received: 08-06-2025
Accepted: 01-09-2025
Introduction
Andrade syndrome, also known as familial amyloid polyneuropathy type I (FAP type I), is a rare autosomal dominant disorder caused by mutations in the transthyretin (TTR) gene(1,2). These mutations result in the misfolding of the transthyretin protein and the deposition of amyloid in various tissues, with a particular affinity for peripheral nerves, although cardiac and gastrointestinal involvement is also common(3).
Clinically, the disease presents as a distal, symmetrical, and progressive sensorimotor polyneuropathy, typically beginning in the feet. Sensory loss, muscle weakness, and impaired balance render the foot especially vulnerable from early stages. The resulting peripheral neuropathy compromises the perception of pain, pressure, and temperature, increasing the risk of unnoticed injuries, persistent infections, and structural deformities.
Unlike other peripheral neuropathies such as diabetic neuropathy or Charcot-Marie-Tooth disease, Andrade syndrome often features more widespread systemic and autonomic dysfunction. This combination heightens the likelihood of severe podiatric complications, including joint instability, chronic ulcers, and destructive arthropathies resembling Charcot foot. Although less frequently reported, these complications can significantly affect gait, balance, and patient independence(4,5).In addition, autonomic impairment reduces vascular regulation and skin integrity, delaying wound healing and increasing susceptibility to plantar ulcers5. Neuropathic pain may appear early, before motor symptoms, progressively impairing physical and emotional well-being(6,7).
Despite advances in treatment—such as tafamidis and patisiran, which have shown efficacy in slowing disease progression(2,8)— the podiatric consequences of the syndrome remain poorly documented. Most available literature focuses on genetic and neurological aspects, with limited attention to the functional implications of foot lesions or to the evaluation of therapeutic and preventive strategies9. Therefore, this systematic review aims to assess the prevalence and clinical features of foot-related complications in patients with Andrade syndrome, to examine how these complications affect mobility and quality of life, and to identify the most common lesions, associated risk factors, and currently applied therapeutic interventions, along with their clinical effectiveness and feasibility.
Materials and methods
This systematic review was conducted in accordance with the PRISMA 2020 (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines(10,11)and was prospectively registered in the PROSPERO database under ID: CRD581310. Among the studies included in this review is the NEURO-Transform trial by Coelho et al. (12), a phase 3 open-label study that provides detailed demographic and clinical data on patients with hereditary transthyretin amyloidosis (ATTRv) related polyneuropathy. This study was selected due to its comprehensive characterization of neuropathic progression and treatment outcomes, which are relevant to the podiatric implications analyzed.
Search strategy
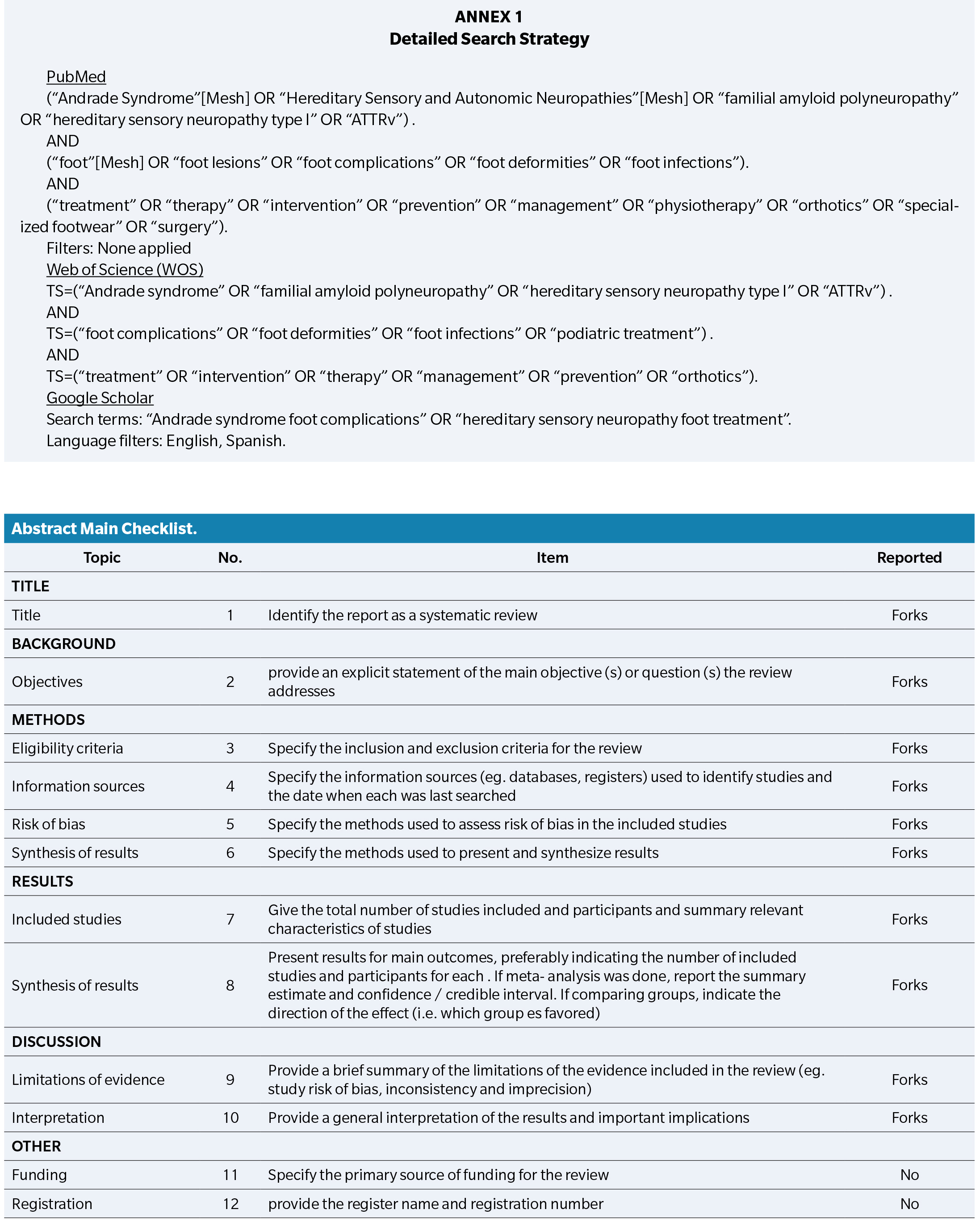
A systematic search was performed in PubMed, Google Scholar and Web of Science (WOS) databases up until 31 December 2024. Google Scholar was included to capture potentially relevant grey literature and sources not indexed in traditional databases.
Following keywords: “Andrade syndrome”, “Hereditary sensory neuropathy type I”, “foot lesions”, “foot complications”, “foot deformities”, “foot infections”, “treatment”, “therapy”, “management”, “intervention”, “prevention”, “specialized footwear”, “physiotherapy”, “surgery”, “orthotics”, “medications”.
The following MeSH terms and keywords were used: “Andrade syndrome”, “Hereditary sensory neuropathy type I”, “foot lesions”, “foot complications”, “foot deformities”, “foot infections”, “treatment”, “therapy”, “management”, “intervention”, “prevention”, “specialized footwear”, “physiotherapy”, “surgery”, “orthotics”, “medications”.
Boolean operators (AND, OR) were used to combine search terms. Search strings were adapted for each database to ensure syntax compatibility.
All articles with Spanish or English abstracts and full texts were assessed. We did not utilize a year limit or filters.
The complete search strategy for each database is provided in Appendix 1.
Inclusion and exclusion criteria
Inclusion criteria consisted in primary research studies published in peer-reviewed journals indexed in PubMed, ScienceDirect, or Google Scholar; studies that investigate the impact of Andrade syndrome/ATTRv on foot health; studies that assess complications, mobility, quality of life, or evaluate preventive or therapeutic interventions; and articles published between 2019 and 2024. Studies in English or Spanish.
Exclusion criteria was review articles, editorials, letters to the editor, conference abstracts, or opinion pieces. Studies not specifically addressing Andrade syndrome or its podiatric implications. Articles published before 2019 or in languages other than English or Spanish. Studies not meeting the quality or methodological standards for systematic inclusion.
Study selection process
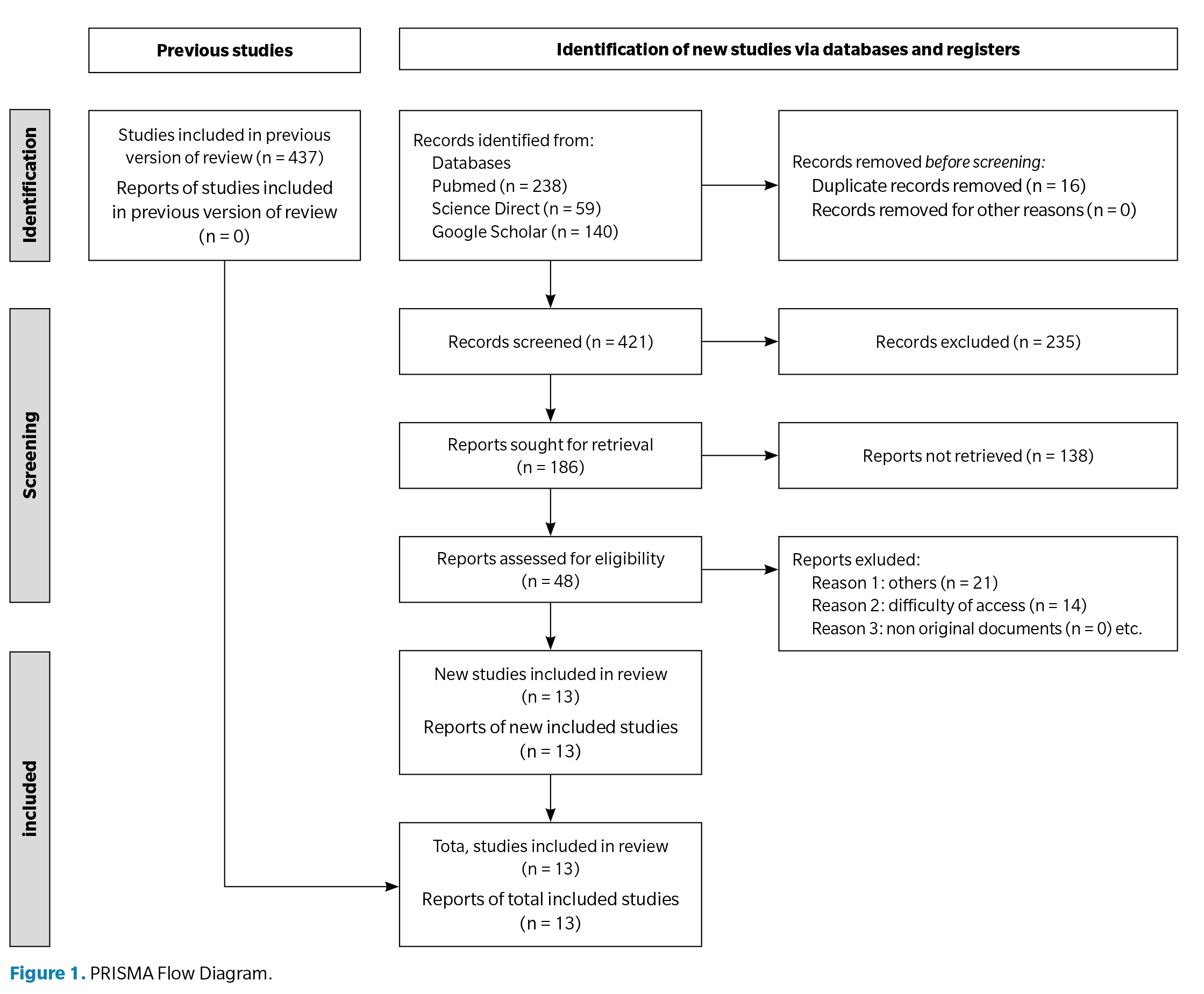
All retrieved records were exported into a reference manager for duplication removal. Two independent reviewers screened titles and abstracts. Full texts were assessed for eligibility based on predefined criteria. Disagreements were resolved by consensus or consultation with a third reviewer. A PRISMA flowchart (Figure 1) summarizes the study selection process, detailing the number of records identified, screened, excluded, and included.
Data extraction and synthesis
Data from the selected studies were systematically extracted into a standardized table, including authors, publication year, study design, population characteristics, intervention/exposure, outcomes, and main findings.
A narrative synthesis approach was applied due to the heterogeneity in study designs and outcome measures. Results were grouped thematically by type of foot complication (injuries, infections, deformities), functional limitations, and impact on quality of life.
Risk of bias assessment
The methodological quality of included studies was evaluated using:
ROBINS-I tool for non-randomized studies.
RoB 2 tool for randomized controlled trials.
The risk of bias was assessed independently by two reviewers. Discrepancies were resolved through discussion.
Results
The diagram illustrating the study selection process for the different stages of the systematic review is shown in Figure 1. Of a total of 499 studies obtained in the initial search, 13 documents met the inclusion criteria and were selected: 3 observational studies, 3 clinical trials, one retrospective study, one expert consensus, one retrospective cohort, and one descriptive cross-sectional study. In addition, 2 additional documents were incorporated from the manual search of reference lists. A table of original articles was compiled with the main characteristics of each study (Table 1). Although thirteen documents were included in the final synthesis, only twelve were original studies with empirical data, while the expert consensus was considered relevant for its clinical contributions.
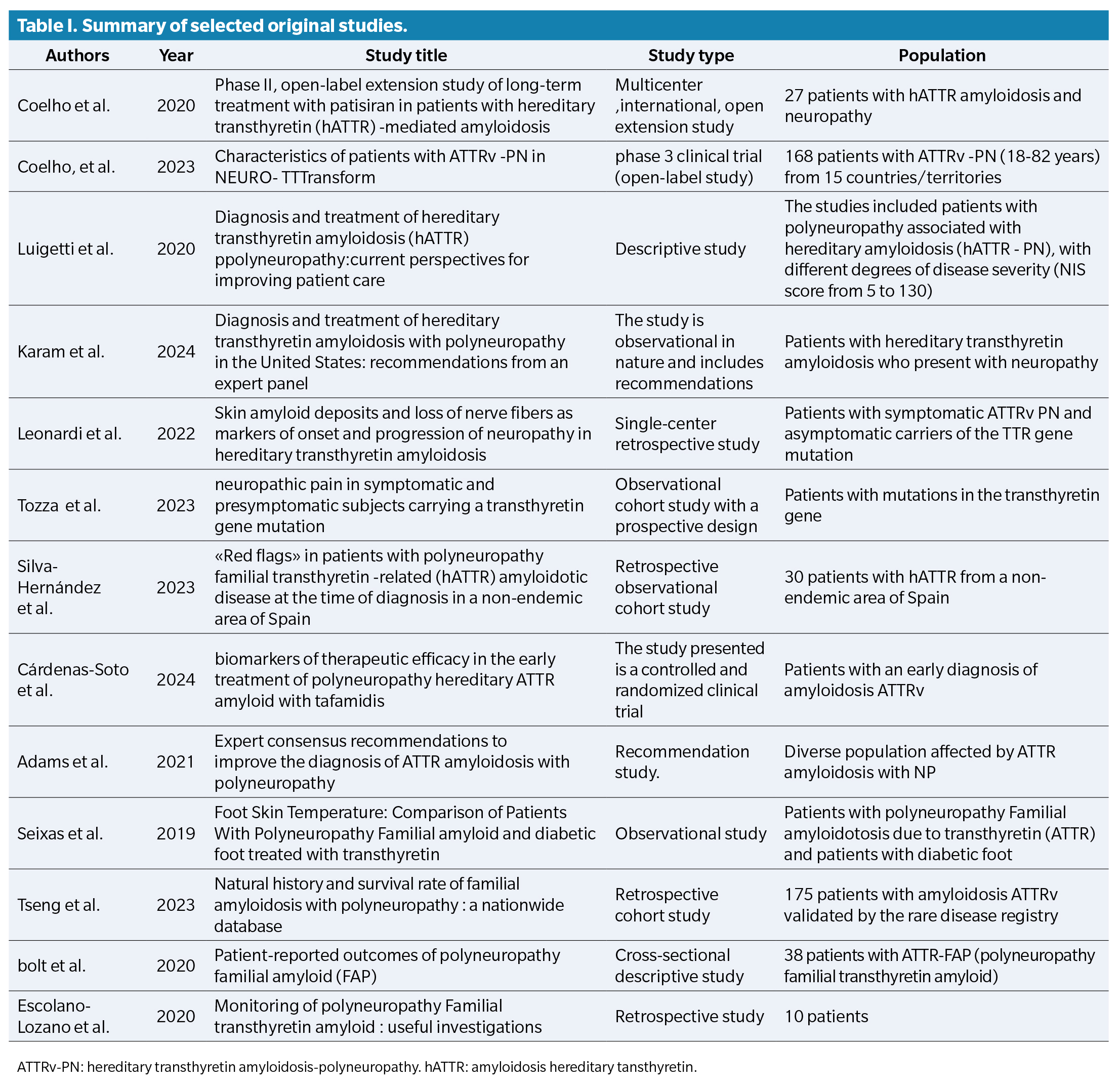
Among the selected studies, several reported specifically on podiatric manifestations of ATTRv, including plantar ulcers, biomechanical deformities, and thermoregulatory changes in the feet. These complications are especially relevant to podiatric practice, as they increase the risk of pressure injuries, gait dysfunction, and reduced autonomy.
Although the article by Karam et al. (2024) was included in the narrative synthesis due to its clinical relevance, it was not assessed using ROBINS-I or RoB 2 tools because it does not report original interventional data. Instead, it represents a panel-based expert consensus aimed at providing clinical recommendations and thus falls outside the scope of bias evaluation tools applied in this review(13).
Risk of bias assessment was performed using two tools: ROBINS-I for non-randomized studies and RoB-2 ( Table 2) for randomized studies ( Table 3).
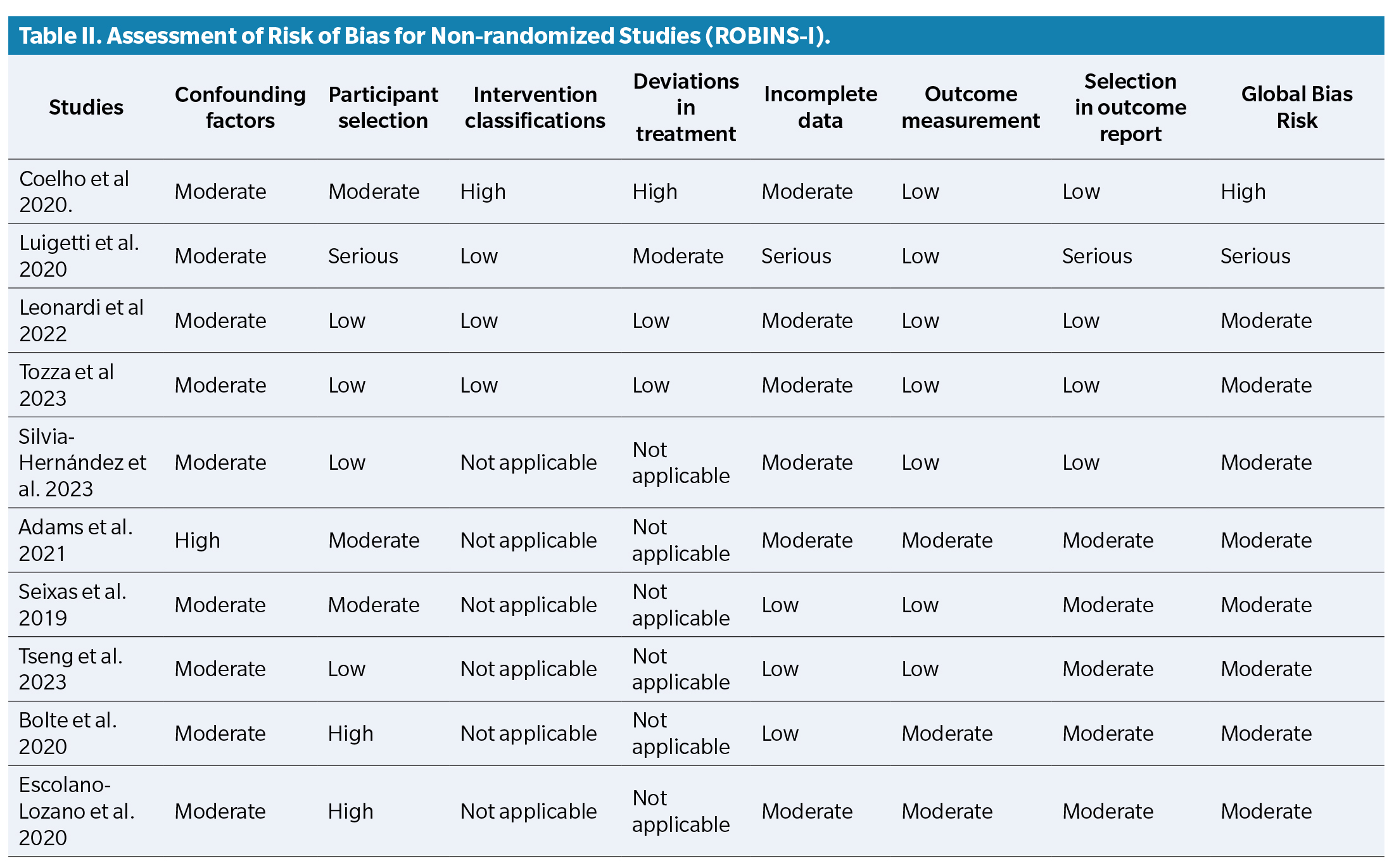
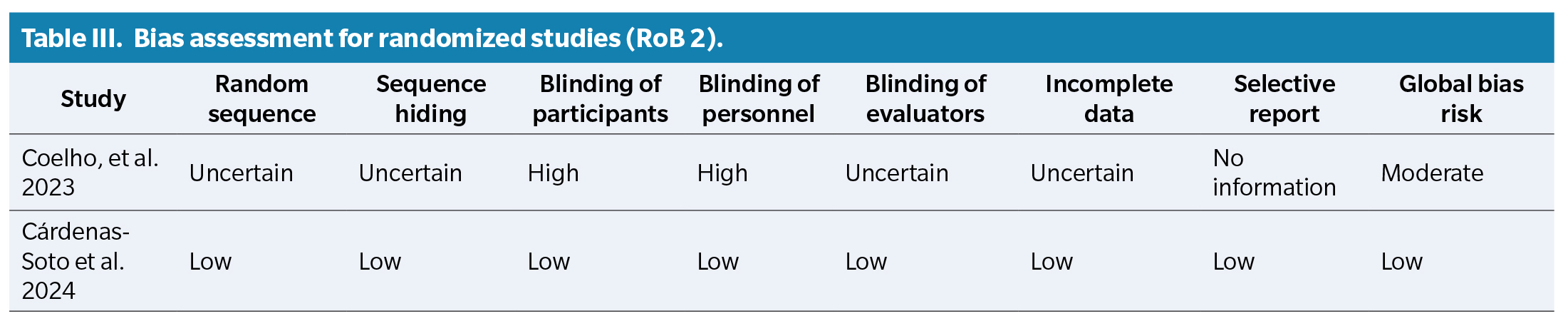
Among the selected studies, several reported specifically on podiatric manifestations of ATTRv, including plantar ulcers, biomechanical deformities, and thermoregulatory changes in the feet. In addition, Leonardi et al. (2022) demonstrated that amyloid deposits in the skin are associated with nerve fiber loss in patients with ATTRv, suggesting their potential value as early markers of neuropathy progression14. Silva-Hernández et al. (2023) emphasized the presence of clinical warning signs at diagnosis in patients from non-endemic areas, underlining the risk of delayed identification(15). Moreover, Cárdenas-Soto et al. (2024) evaluated cutaneous biomarkers that may guide early therapeutic decisions and improve monitoring of disease progression, with implications for foot health(16). These complications are especially relevant to podiatric practice, as they increase the risk of pressure injuries, gait dysfunction, and reduced autonomy.
Familial amyloid polyneuropathy (ATTR) is correlated with a significant prevalence of foot-related complications, including neuropathy and skin temperature disturbances, which may lead to vasoconstriction and decreased sensory perception (Luigetti et al.; Bolte et al.) (17,18). While similar complications are observed in diabetic and Charcot-Marie-Tooth neuropathies, the systemic nature and autonomic involvement of ATTRv make its podiatric impact more insidious and often underdiagnosed. This underscores the need for targeted foot care strategies tailored to this population.
In individuals diagnosed with ATTR, the observed decrease in sympathetic nerve innervation has been shown to exacerbate these complications, particularly in patients with concomitant diabetes mellitus (Lepesis et al.) (19). A study by Leonardi et al. (14) revealed that 80 % of symptomatic individuals presenting with hereditary amyloidosis-related neuropathy showed amyloid deposits in the dermis, which may contribute to nerve fiber degeneration and consequently complications such as foot lesions and deformities. Furthermore, research by Karam et al. (2024) (13) karam emphasizes that autonomic neuropathy, which can present as disturbances in gastrointestinal function and associated symptoms, also negatively affects foot health, leading to further complications. Furthermore, research by Tseng et al. (2023)20 documented that individuals with ATTRV had a higher incidence of foot complications compared to individuals without this condition, underscoring the need for close follow-up.
Research has shown that mobility is significantly compromised in individuals diagnosed with ATTR, negatively impacting their overall quality of life (Adams et al.)3. Research conducted by Coello et al. (21) focused on assessing the impact of neuropathy on the quality of life of individuals suffering from FAP. The results revealed that patients experience a significant impairment in their quality of life, associated with manifestations such as neuropathic pain, muscle weakness, and mobility problems. Furthermore, autonomic neuropathy, which can manifest through gastrointestinal and cardiovascular symptoms, was also observed to play a role in the impairment of quality of life. In a cross-sectional descriptive analysis by Bolte et al. (18), evidence was if individuals with familial amyloid polyneuropathy reported reduced quality of life related to decreased mobility and foot discomfort. Furthermore, research by Karam et al. (13) indicates that confirmation of ATTR diagnosis can be achieved through biopsies and imaging, facilitating more effective treatment and potentially improved quality of life. Furthermore, the randomized controlled trial by Lepesis et al. (19) illustrated that foot and ankle mobilizations coupled with home-based stretching improved mobility in patients affected by diabetic neuropathy, implying that analogous strategies may be beneficial for individuals with ATTR. The assessment of autonomic function using instruments such as Sudoscan has proven advantageous in delineating sensory abnormalities in the feet, which are intrinsically related to the quality of life of these patients (Luigetti et al.; Bolte et al.) (2,18).
Coelho et al. (21) concluded that appropriate therapeutic interventions can have a favorable effect on disease progression and improve the quality of life of patients with PAF. The importance of early diagnosis and timely treatment to optimize therapeutic outcomes was emphasized. Research by Tozza et al. (6) emphasizes the high prevalence and early onset of neuropathic pain in individuals with transthyretin gene mutations. The 2023 study specifically showed that pain is frequently present even in presymptomatic carriers, supporting the need for early sensory assessment and preventive podiatric care. Neuropathic pain can restrict patients’ ability to perform physical activities and, consequently, lead to an overall reduction in mobility. Restricting free movement not only impairs physical autonomy but can also contribute to a cycle of inactivity that exacerbates neuropathic symptoms and muscle weakness. Patients’ quality of life is compromised not only by physical discomfort but also by mobility limitations. The inability to perform daily, occupational, or recreational activities can lead to feelings of frustration, isolation, and depression. Research postulates that effective treatment of neuropathic pain is essential to improving quality of life, as pain relief allows patients to regain a degree of mobility and, consequently, their independence and overall well-being.
Research conducted by Silva-Hernández et al. (15) discusses the implications of neuropathy linked to hereditary amyloidosis on patients’ mobility. Manifestations such as muscle weakness and balance disturbances can impede people’s ability to navigate their environments safely and efficiently. This not only increases the likelihood of falls and subsequent injuries but also leads to greater dependence on others for routine tasks, which compromises patient autonomy. Quality of life is profoundly influenced by mobility limitations. Research emphasizes that the quality of life of these individuals is not only assessed from the perspective of physical health, but also through their ability to participate in social interactions and family activities. Mobility is a fundamental element of quality of life, and its decline can have adverse effects on patients’ motional and social well-being.
Therapeutic modalities include foot and ankle mobilizations along with stretching exercises, which have demonstrated favorable results regarding mobility in individuals diagnosed with diabetic peripheral neuropathy (Lepesis et al.) (19). Furthermore, it is postulated that an expansion of research focused on specific therapeutic interventions could improve patient care and mitigate foot-related complications (Luigetti et al.; Adams et al.) (2,3).
The study conducted by Seixas et al. (5), entitled “Effects of Hereditary Transthyretin Amyloidosis on Patients’ Quality of Life,” examines the impact of hereditary transthyretin amyloidosis (HTTA) on the quality of life of affected patients. The study’s main findings: The studyincludess that individuals with HTTA experience substantial impairment in their quality of life, attributable to symptoms associated with the disorder, which include neuropathic pain, muscle weakness, and autonomic dysfunction. The analysis identifies prevalent symptoms that negatively impact quality of life, including gastrointestinal disorders, mobility impairments, and cardiovascular manifestations. These symptoms not only negatively affect physical health but also have emotional and social ramifications. According to patient reports, a significant association is established between symptom severity and quality of life. As symptom intensity increases, there is a concomitant decline in quality of life, underscoring the need for effective symptom management. Research underscores the importance of timely and appropriate treatment to improve patients’ quality of life. It is proposed that a holistic approach to disease management, incorporating both pharmacological interventions and psychological and social support, is essential to improving patients’ overall well-being. This study of Seixas et al. (5) clarifies the profound effects of hereditary transthyretin amyloidosis on patients’ daily lives. This emphasizes the need for multidisciplinary strategies in the treatment of the disorder, which should focus not only on physical aspects but also on emotional and social dimensions relevant to quality of life.
Research conducted by Cárdenas-Soto et al. (16) focused on evaluating the therapeutic efficacy of tafamidis in individuals with a preliminary diagnosis of hereditary transthyretin amyloidosis (ATTRV). This study indicates that administering tafamidis during the early stages of amyloid polyneuropathy may improve clinical outcomes and underscores the need to further explore biomarkers that may facilitate treatment personalization and monitoring of therapeutic efficacy. This research reveals that tafamidis intervention is effective in slowing the progression of neuropathy compared to a control cohort. This finding implies that tafamidis could serve as a beneficial therapeutic option for patients in the early stages of the condition. Skin biomarkers are also studied as potential indicators of therapeutic efficacy. These biomarkers have the potential to provide information on treatment responses and disease progression, vital elements for the clinical monitoring of amyloidosis. The study underscores the critical role of early diagnosis of ATTRV amyloidosis, as prompt intervention with tafamidis can significantly influence both quality of life and the trajectory of disease progression.
The research conducted by Escolano-Lozano et al. (17), entitled “Evaluation of Neuropathy in Patients with Hereditary Transthyretin Amyloidosis: A Multidimensional Approach”, focuses on the assessment and treatment of neuropathy in individuals diagnosed with hereditary transthyretin amyloidosis (ATTRV).
Research advocates for a multidimensional approach to neuropathy assessment, incorporating both clinical evaluations and the use of sophisticated diagnostic tools. This methodology facilitates a more complete characterization of neuropathy in the aforementioned patients.
The study conducted by Escolano-Lozano et al. (17) clarifies the intricate nature of neuropathy in hereditary transthyretin amyloidosis and the need for a holistic approach to its assessment and treatment. By addressing the clinical and functional dimensions of neuropathy, patients‘ quality of life can be improved and treatment outcomes optimized.
These studies support the initial research aim of this systematic review: to determine the extent to which foot complications in ATTRv patients affect mobility and quality of life. The evidence highlights the need for multidisciplinary podiatric involvement, particularly in the early identification and management of neuropathic pain, deformities, and autonomic dysfunction in the lower limbs.
Discussion
This systematic review demonstrates that Andrade syndrome (familial amyloid polyneuropathy type I) has a significant impact on foot health, comparable in severity, though not in prevalence, to other well-studied neuropathies such as diabetes and Charcot-Marie-Tooth disease(4,7,10). The progressive sensory loss, combined with the autonomic dysfunction characteristic of this condition, increases vulnerability to foot injuries, ulcerations, and infections, all of which directly compromise quality of life and functional independence(5,14,17).
Unlike more prevalent peripheral neuropathies, such as diabetic neuropathy, where the incidence of Charcot foot or plantar ulcers is well documented, podiatric complications in Andrade syndrome have been insufficiently described(6,10). Nevertheless, several studies included in this review (e.g., Coelho et al., Leonardi et al., Silva-Hernández et al.) confirm that sensory deficits and amyloid infiltration in the dermis and nerve fibers may lead to biomechanical alterations of similar magnitude(12,14,15).
From a functional perspective, most studies consistently report a progressive decline in mobility, particularly in the advanced stages of the disease, which negatively affects patients’ ability to carry out daily living activities(13,18,20). This functional impact has been quantified using quality-of-life scales (such as EQ-5D or Norfolk QoL-DN), showing high levels of disability compared to other hereditary neuropathies(11,18,21). Moreover, the psychological burden, including fear of injury and social isolation, adds further complexity to patient care.
Although disease-modifying treatments such as tafamidis and patisiran have demonstrated efficacy in slowing neurological progression(2,8,12,16), the reviewed studies reveal a lack of podiatry-focused strategies for preventing structural foot complications or restoring locomotor function(17,19). Interventions including physiotherapy, customized orthotics, foot health education, and targeted pain management may play a key role in comprehensive care(5,13,19).
The foot emerges as one of the most affected anatomical structures in Andrade syndrome due to the distal distribution of neuropathy. This review underscores the need to integrate specialized podiatric evaluation as an essential component of comprehensive disease management. Multidisciplinary care should include not only strategies aimed at slowing neurological progression but also measures to prevent structural complications, preserve mobility, and maintain functional autonomy. Strengthening this clinical approach could improve therapeutic outcomes and the overall quality of life for individuals affected by this condition(2,8,12,13,19).
The primary limitation of this review is the limited number of studies directly addressing foot involvement in Andrade syndrome. Many of the included articles focus broadly on neurological or genetic aspects without incorporating specific podiatric variables. Furthermore, methodological heterogeneity, variability in outcome measures, and small sample sizes in some studies hinder robust quantitative synthesis.
From the results of this review several clinical and research recommendations can be made such us conduct clinical studies focused on the foot as a key functional unit in disability progression among ATTRv patients, systematically incorporate podiatric assessment into multidisciplinary disease monitoring, investigate the effectiveness of interventions such as joint mobilization, orthopedic footwear, or thermographic sensors for early detection of foot injuries(5,13,19) and establish foot screening protocols, particularly in non-endemic regions, where diagnosis is often delayed(15, 16).
Conclusions
This systematic review confirms that familial amyloid polyneuropathy (FAP), also known as hereditary transthyretin amyloidosis (ATTRv), has a significant impact on foot health, primarily due to the progressive sensory and autonomic neuropathy characteristic of this disease. Patients experience a marked reduction in pain and temperature perception in the distal extremities, increasing the risk of undetected injuries, ulcerations, and biomechanical deformities.
These complications severely affect gait, balance, and functional autonomy, contributing to progressive mobility loss and a substantial decline in quality of life. Neuropathic pain and delayed wound healing particularly exacerbate disability, especially in advanced stages of the disease.
Although disease-modifying therapies such as tafamidis and patisiran have shown efficacy in slowing neurological progression, the reviewed literature reveals a clear lack of strategies focused on the prevention and treatment of podiatric complications. Few studies have addressed interventions such as foot orthotics, physiotherapy, or self-care education, despite their potential clinical benefits.
The development of a care model that incorporates specialized podiatric assessment as part of multidisciplinary follow-up is essential. Moreover, further research is needed to evaluate the effectiveness of specific interventions aimed at preserving functionality, reducing foot-related morbidity, and improving the quality of life of individuals affected by Andrade syndrome.
Conflict of interest
The authors declare that there are no conflicts of interest related to the conduct and publication of this study
Funding
None
Contribution of authors
Conception and design of the study: MPAG; ATG; SHR. Literature search/Collection and search of studies: MPAG; BBF: SHR. Analysis and interpretation of results: BBF; MMA. Creation, writing, and preparation of draft: AGL; BBF; MPAG. Final review: SHR; BBF; MPAG
Supplement material
Annex 1
References
- Murakami T, Yokoyama T, Mizuguchi M, Toné S, Takaku S, Sango K, et al. A low amyloidogenic E61K transthyretin mutation may cause familial amyloid polyneuropathy. J Neurochem. 2021;156(6):957-66. DOI: 10.1111/jnc.15162.
- Luigetti M, Romano A, di Paolantonio A, Bisogni G, Sabatelli M. Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: Current perspectives on improving patient care. Ther Clin Risk Manag. 2020;16:109-23. DOI:10.2147/TCRM.S219979.
- Adams D, Ando Y, Beirão JM, Coelho T, Gertz MA, Gillmore JD, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109-22. DOI:10.1007/s00415-019-09688-0.
- Kucera T, Shaikh HH, Sponer P. Charcot neuropathic arthropathy of the foot: A literature review and single-center experience. J Diabetes Res. 2016;2016:3207043. DOI:10.1155/2016/3207043.
- Seixas A, Vilas-Boas MC, Carvalho R, Coelho T, Ammer K Vilas-Boas JP, et al. Skin temperature of the foot: Comparing transthyretin familial amyloid polyneuropathy and diabetic foot patients. Comput Methods Biomech Biomed Eng Imaging Vis. 2019;7(5-6):504-11. DOI:10.1080/21681163.2018.1471621.
- Tozza S, Luigetti M, Antonini G, Mazzeo A Severi D, di Paolantonio A, et al. Neuropathic pain experience in symptomatic and presymptomatic subject carrying a transthyretin gene mutations. Front Neurol. 2023;14:1109782. DOI:10.3389/fneur.2023.1109782.
- Girach A, Julian TH, Varrassi G, Paladini A, Vadalouka A, Zis P. Quality of life in painful peripheral neuropathies: A systematic review. Pain Res Manag. 2019;2019:2091960. DOI:10.1155/2019/2091960.
- Coelho T, Maia LF, Martins da Silva A, Cruz MW, Palnté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatmen of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14. DOI: 10.1007/s00415-013-7051-7.
- Ziegler D, Burow S, Landgraf R, Lobmann R, Reiners K, Rett K, et al. Current practice of podiatrists in testing for diabetic polyneuropathy and implementing foot care (PROTECT Study Survey 2). Endocr Pract. 2024;30(9):817-21. DOI:10.1016/j.eprac.2024.06.006.
- Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. DOI:10.1371/journal.pmed.1000097.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. DOI:10.1136/bmj.n71.
- Coelho T, Waddington Cruz M, Chao CC, Parman Y, Wixner J, Weiler M, et al. Characteristics of patients with hereditary transthyretin amyloidosis-polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an open-label phase 3 study of eplontersen. Neurol Ther. 2023;12(1):267-87. DOI: 10.1007/s40120-022-00414-z.
- Karam C, Mauermann ML, Gonzalez-Duarte A, Kaku MC, Ajroud-Driss S, Brannagan TH, et al. Diagnosis and treatment of hereditary transthyretin amyloidosis with polyneuropathy in the United States: Recommendations from a panel of experts. Muscle Nerve. 2024;69(3):273-87. DOI: 10.1002/mus.28026.
- Leonardi L, Adam C, Beaudonnet G, Beauvais D, Cauquil C, Not A, et al. Skin amyloid deposits and nerve fiber loss as markers of neuropathy onset and progression in hereditary transthyretin amyloidosis. Eur J Neurol. 2022;29(5):1477-87. DOI: 10.1111/ene.15268.
- Silva-Hernández L, Horga Hernández A, Valls Carbó A, Guerrero Sola A, Montalvo-Moraleda MT, Galán Dávila L. Red flags in patients with hereditary transthyretin amyloidosis at diagnosis in a non-endemic area of Spain. Neurology (Eng Ed). 2023;38(2):87-92. DOI: 10.1016/j.nrl.2020.06.009.
- Cárdenas-Soto K, Dominguez X, Cortes G, Tsai F, Saniger MDM, Guraieb-Chahin P, et al. Cutaneous biomarkers of therapeutic efficacy in early treatment of hereditary ATTR amyloid polyneuropathy with tafamidis. J Peripheral Nervous Sys. 2024;29(2):221-31. DOI: 10.1111/jns.12624.
- Escolano-Lozano F, Geber C, Barreiros AP, Birklein F. Follow-up in transthyretin familial amyloid polyneuropathy: Useful investigations. J Neurol Sci. 2020; 413:116776. DOI: 10.1016/j.jns.2020.116776.
- Bolte FJ, Langenstroer C, Friebel F, Hüsing-Kabar A, Dugas M, Schmidt HH. Patient-reported outcomes on familial amyloid polyneuropathy (FAP). Orphanet J Rare Dis. 2020;15(1):287. DOI: 10.1186/s13023-020-01575-6.
- Lepesis V, Paton J, Rickard A, Latour JM, Marsden J. Effects of foot and ankle mobilisations combined with home stretches in people with diabetic peripheral neuropathy: A proof-of-concept RCT. J Foot Ankle Res. 2023;16(1):88. DOI: 10.1186/s13047-023-00690-4.
- Tseng W, Huang H, Li C, Chang C, Chan WP, Lin K, et al. Natural history and survival rate of familial amyloidosis with polyneuropathy: A nationwide databank. Ann Clin Transl Neurol. 2023;10(5):779-86. DOI: 10.1002/acn3.51765.
- Coelho T, Adams D, Conceição I, Waddington-Cruz M, Schmidt HH, Buades J, et al. A phase II, open-label, extension study of long-term patisirán treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. DOI: 10.1186/s13023-020-01399-4.
Annex